=============================================================================================
tract_van.sh
(c) Michael Hart, University of British Columbia, August 2020
Co-developed with Dr Rafael Romero-Garcia, University of Cambridge
Function to run tractography on clinical DBS data (e.g. from UBC Functional Neurosurgery Programme)
Based on the following data: GE scanner, 3 Tesla, 32 Direction DTI protocol
Example:
tract_van.sh --T1=mprage.nii.gz --data=diffusion.nii.gz --bvecs=bvecs.txt --bvals=bvals.txt
Options:
Mandatory
--T1 structural (T1) image
--data diffusion data (e.g. standard = single B0 as first volume)
--bvecs bvecs file
--bvals bvals file
Optional
--acqparams acquisition parameters (custom values, for Eddy/TopUp, or leave acqparams.txt in basedir)
--index diffusion PE directions (custom values, for Eddy/TopUp, or leave index.txt in basedir)
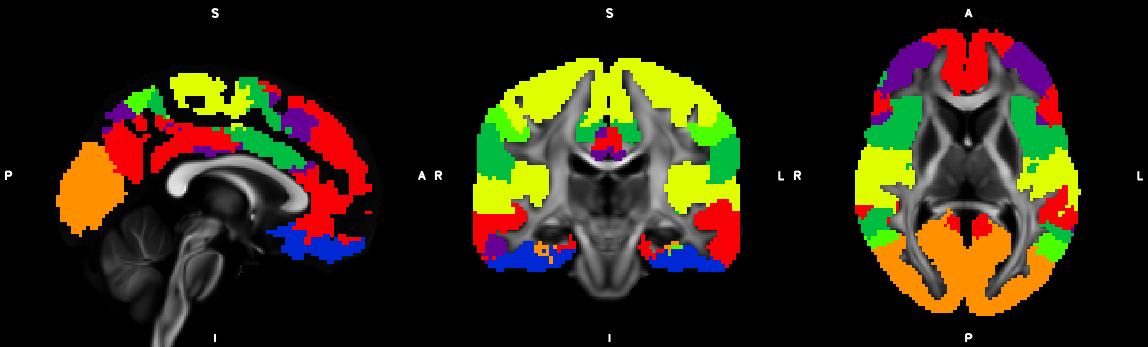
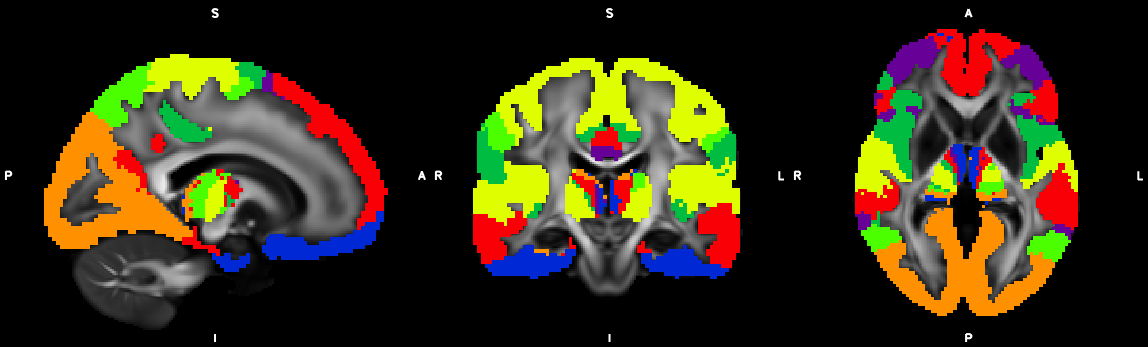
--segmentation additional segmentation template (for segmentation: default is Yeo7)
--parcellation additional parcellation template (for connectomics: default is AAL90 cortical)
--nsamples number of samples for tractography (xtract, segmentation, connectome)
-d denoise: runs topup & eddy (see code for default acqparams/index parameters or enter custom as above)
-p parallel processing (slurm)*
-o overwrite
-h show this help
-v verbose
Pipeline
1. Baseline quality control
2. FSL_anat*
3. Freesurfer*
4. De-noising with topup & eddy - optional (see code)
5. FDT pipeline
6. BedPostX*
7. Registration
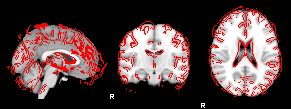
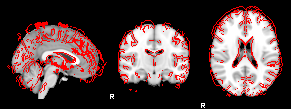
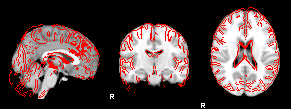
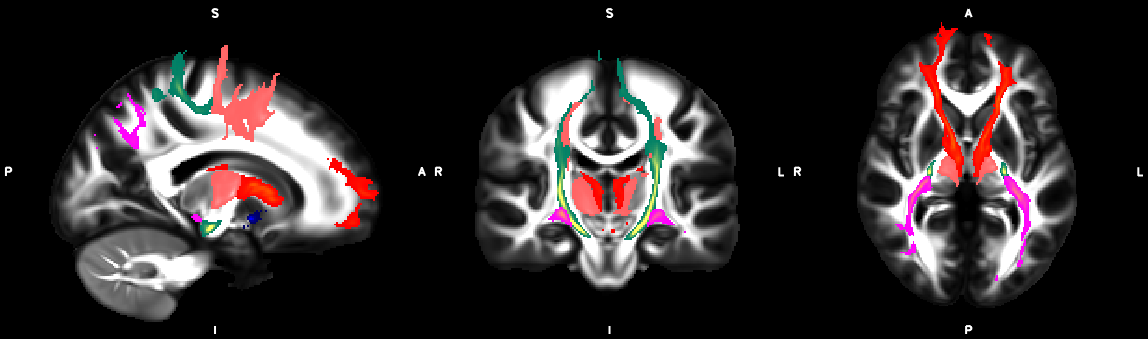
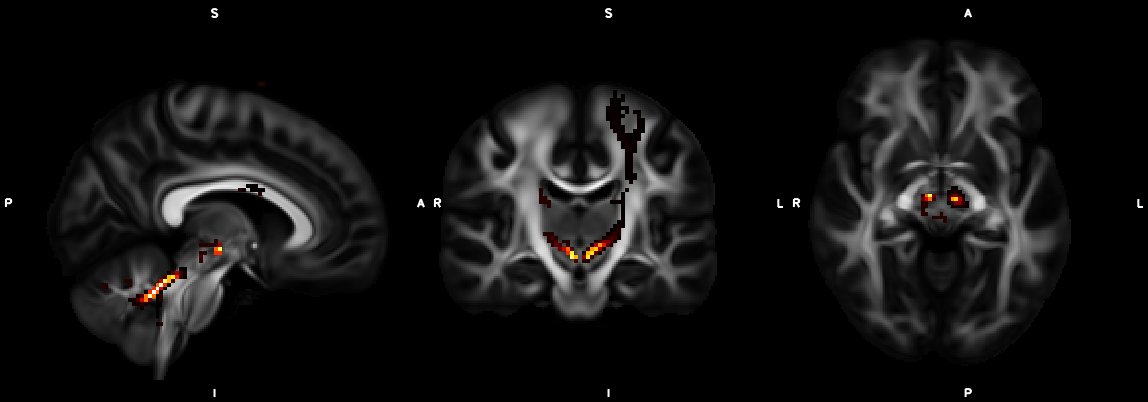
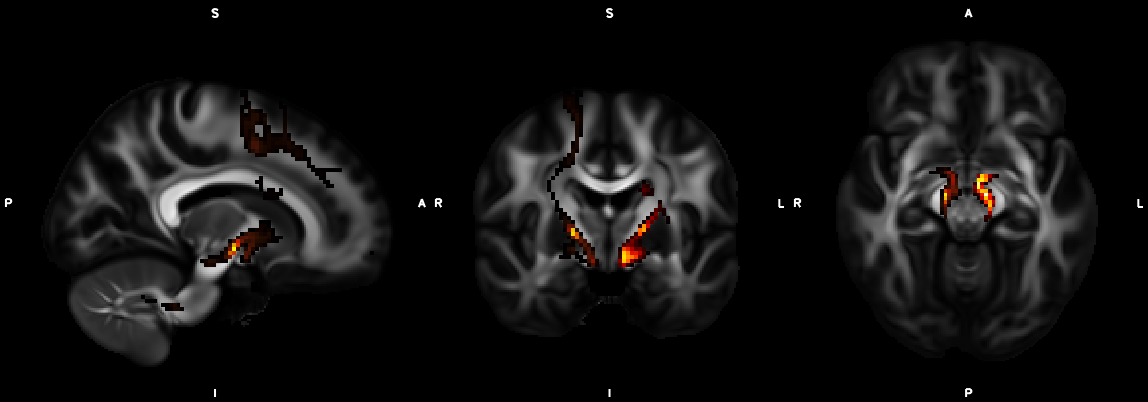
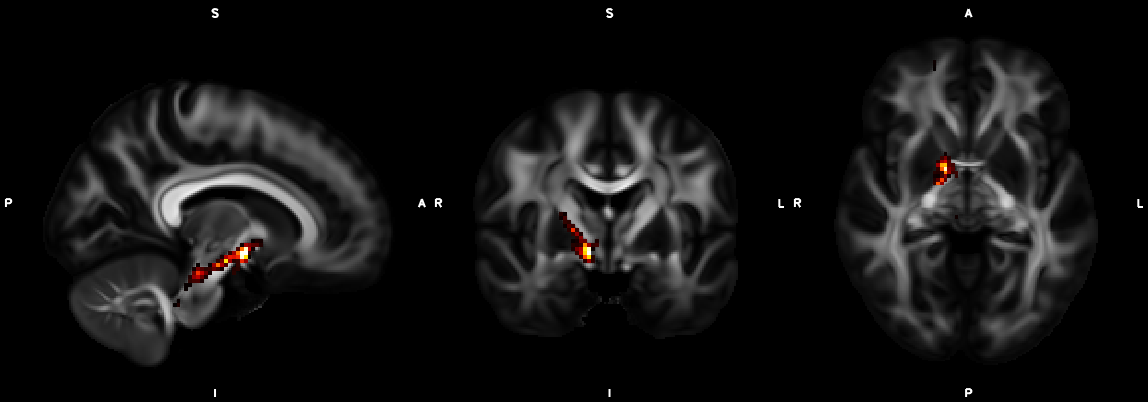
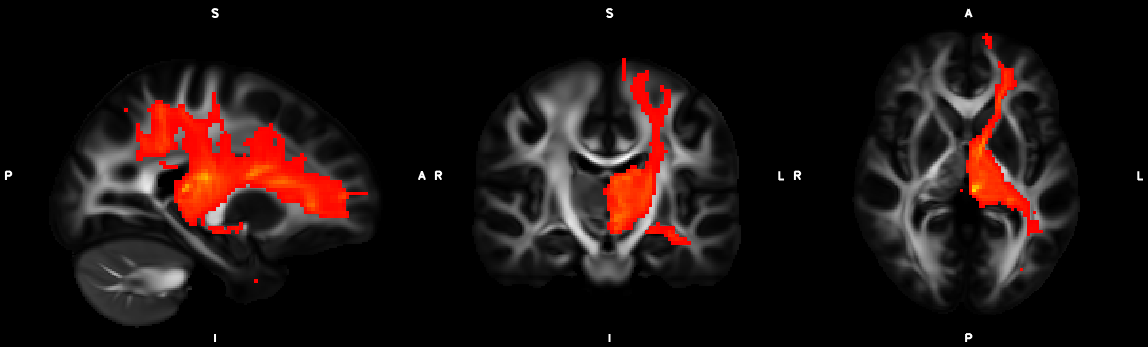
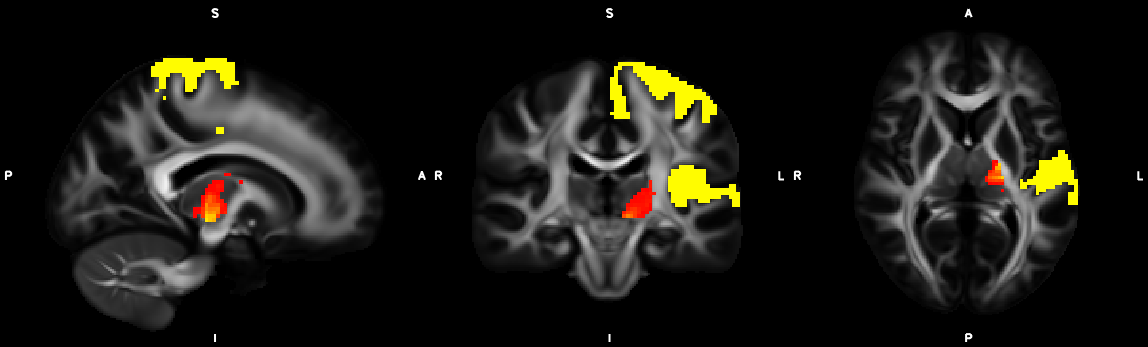
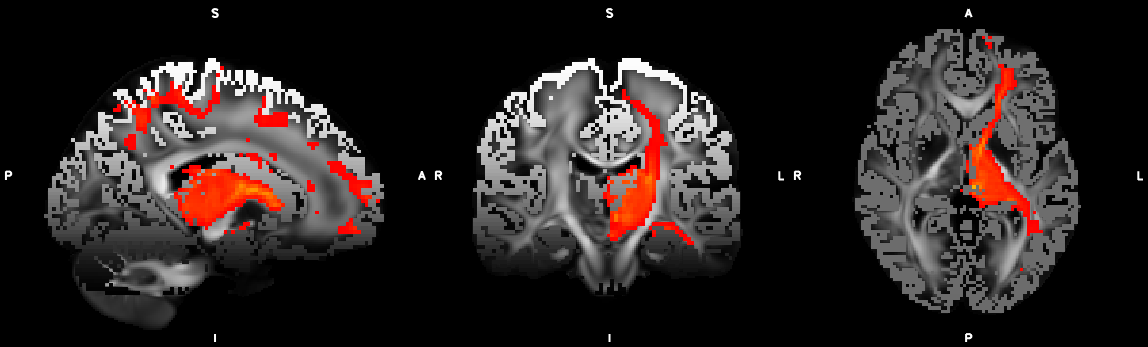
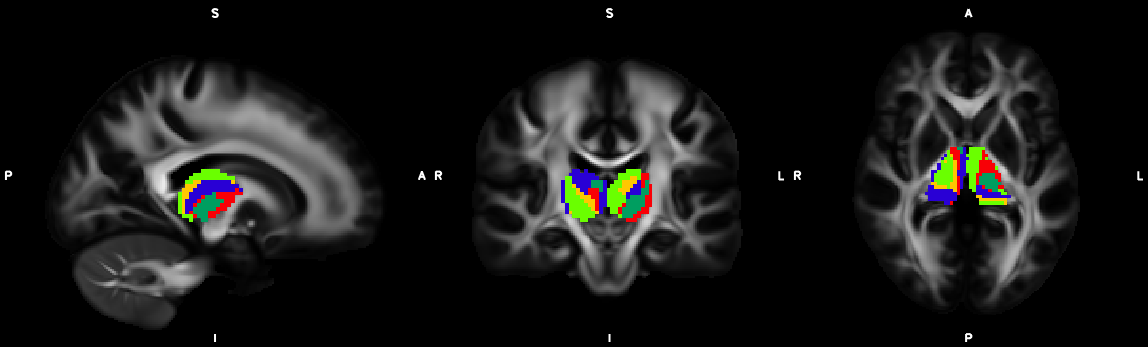
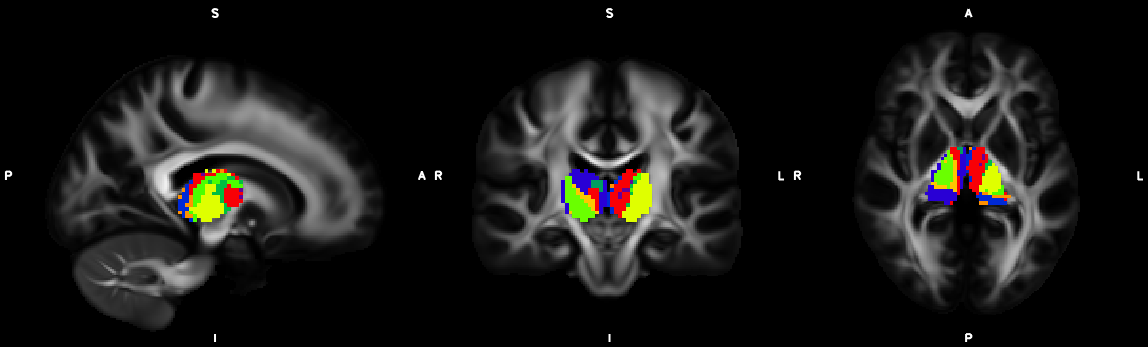
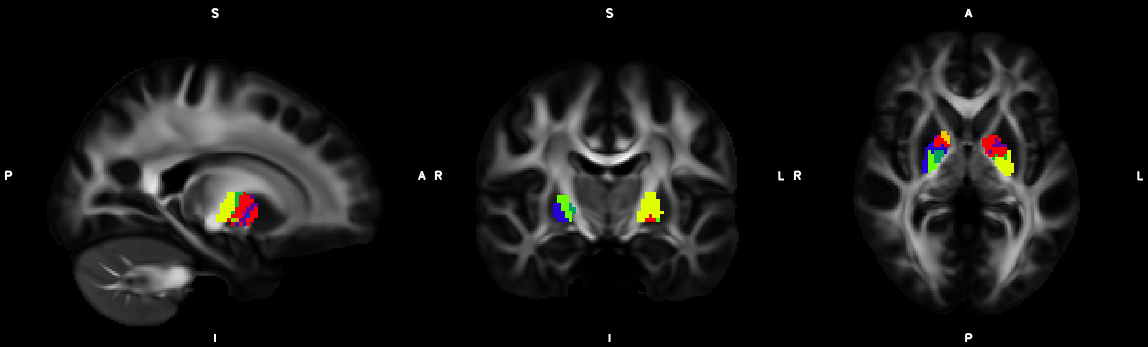
8. XTRACT (including custom DBS tracts)
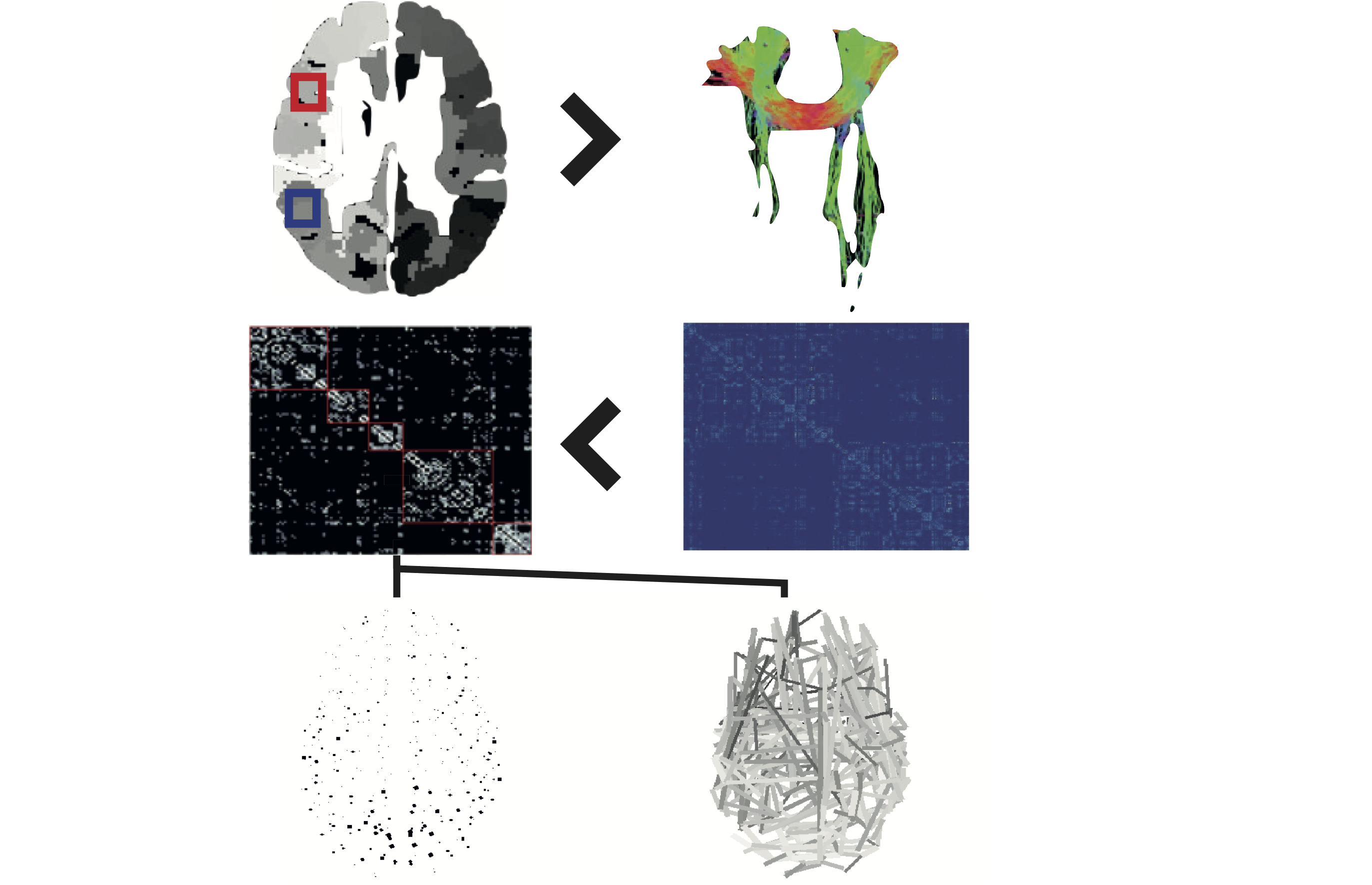
9. Segmentation (probtrackx2)
10. Connectomics (probtrackx2)*
Version: 1.0
History: original
NB: requires Matlab, Freesurfer, FSL, ANTs, and set path to codedir
NNB: SGE / GPU acceleration - change eddy, bedpostx, probtrackx2, and XTRACT calls
=============================================================================================